
—
Molecular Construction: Advanced Methodologies in Solid-Phase Peptide Synthesis (SPPS) and Purification
Introduction to Synthetic Peptide Chemistry
The ability to synthesize specific sequences of amino acids reliably and efficiently has revolutionized biochemical research, pharmacology, and structural biology. While biological expression systems (recombinant DNA technology) are ideal for synthesizing massive protein structures, chemical synthesis provides paramount control for generating short to medium-length peptides (typically 2 to 100 amino acids). Chemical synthesis allows for precise incorporation of unnatural or modified amino acids (D-isoforms, fluorophores, lipid conjugates), specific isotopic labeling, and complex structural modifications (cyclization, pegylation) that are intrinsically difficult or entirely impossible using biological systems. The cornerstone of modern in-vitro peptide production is Solid-Phase Peptide Synthesis (SPPS). This comprehensive review details the intricate chemistry, sequential methodologies, coupling optimization, and critical analytical purification protocols that constitute contemporary SPPS.
The Paradigm Shift: Bruce Merrifield and the Solid Support
Before the 1960s, peptides were synthesized using classic liquid-phase methods. This approach was excruciatingly slow, incredibly inefficient, and prone to massive yield losses at each step due to the necessity of purifying the intermediate growing peptide after every single amino acid addition.
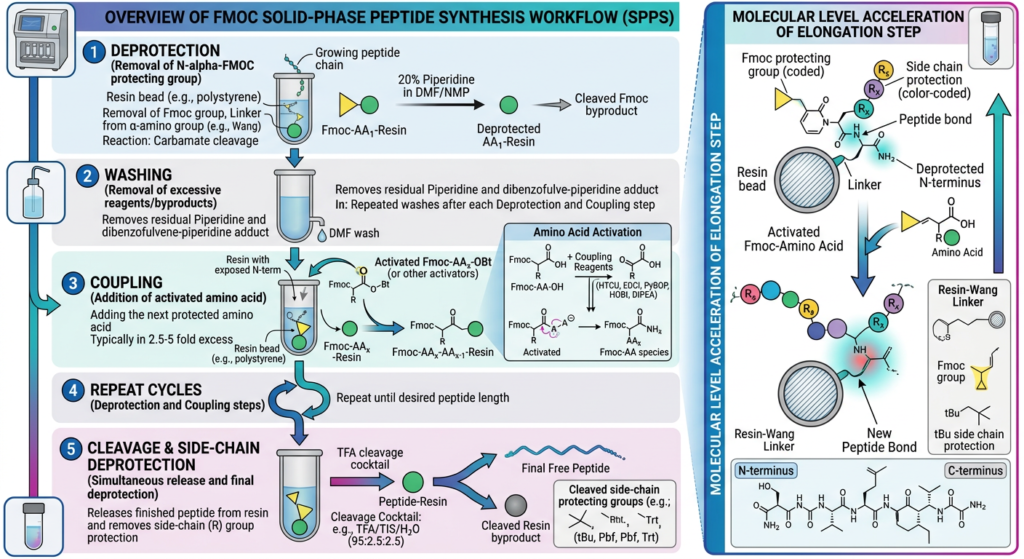
In 1963, R. Bruce Merrifield published a revolutionary paradigm that earned him the Nobel Prize in Chemistry: Solid-Phase Peptide Synthesis. The absolute core ingenuity of SPPS lies in anchoring the C-terminal amino acid of the desired sequence to an insoluble solid matrix (typically a porous polymeric resin bead). Because the growing peptide chain remains covalently immobilized to the resin throughout the synthesis, all unreacted reagents, excess amino acids, and soluble byproducts can be completely removed simply by washing and filtering the resin beads. This eliminated the need for complex intermediate purifications, enabling a rapid, repetitive, and highly automated cyclical process. Synthesis inherently proceeds in a specific direction: from the C-terminus (carboxyl end) toward the N-terminus (amino end), the reverse of biological ribosomal translation.
The Chemistry of Fmoc-SPPS: A Cyclical Process
Contemporary peptide synthesis predominantly utilizes the “Fmoc/tBu” methodology, largely replacing the older, harsher “Boc/Bzl” strategy which required dangerous hydrofluoric acid for final cleavage. The Fmoc approach relies on orthogonal protecting groups—groups that can be removed under entirely different, non-interfering chemical conditions.
1. Protection Strategies
To ensure that only the desired peptide bond initiates, reactive functional groups must be masked.
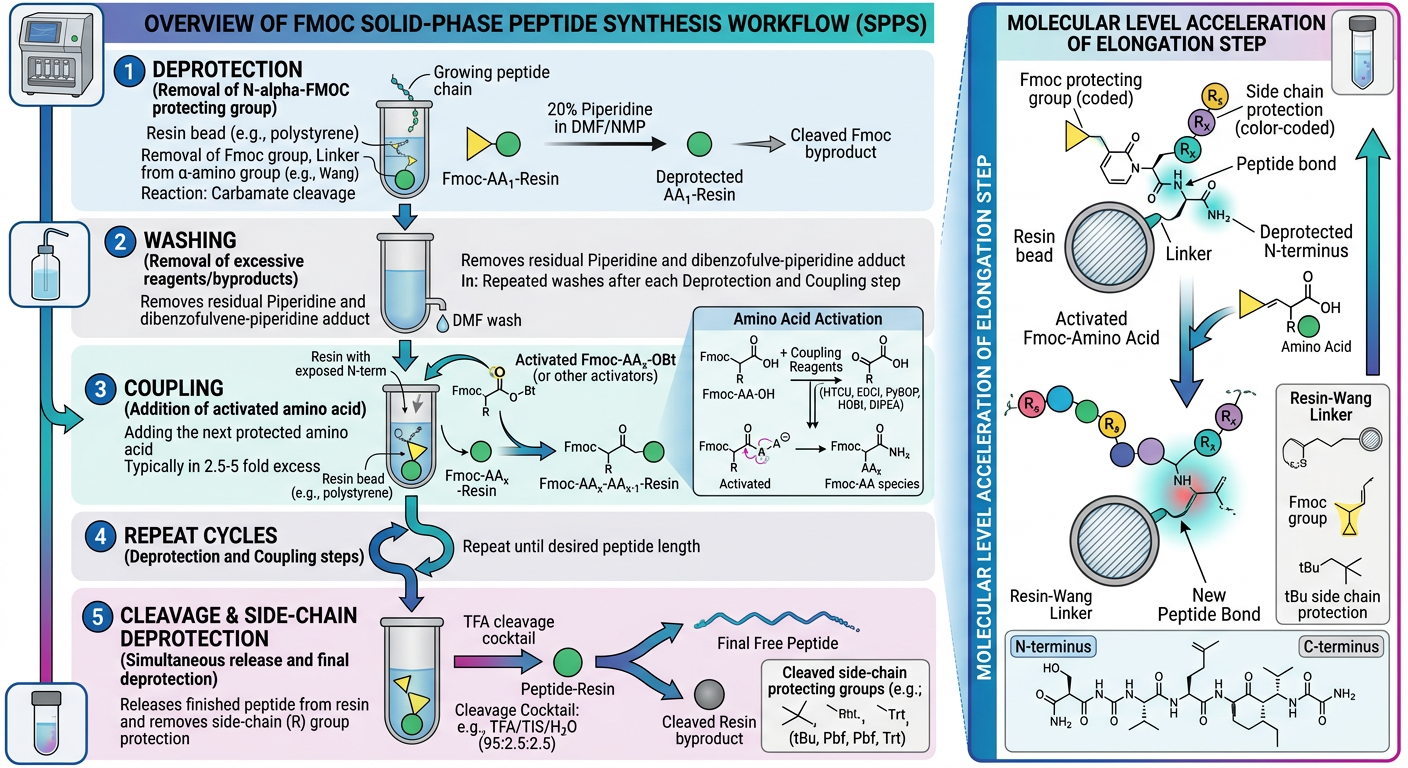
* Targeting the Alpha-Amino Group ($alpha$-$NH_2$): The critical N-terminal amino group of each incoming amino acid is transiently protected by a 9-fluorenylmethyloxycarbonyl (Fmoc) group. The Fmoc group is highly stable to strongly acidic conditions but is rapidly and quantitatively labile to mild organic bases (typically 20% Piperidine in Dimethylformamide, DMF).
* Targeting Side-Chains: Reactive side chains of specific amino acids (e.g., the thiol of Cysteine, the hydroxyl of Serine, the epsilon-amino group of Lysine) are permanently protected using groups like tert-butyl (tBu), trityl (Trt), or pentamethyldihydrobenzofuran-5-sulfonyl (Pbf). These bulky side-chain protecting groups are remarkably resistant to the basic conditions used for Fmoc removal but are highly sensitive to strong acids (specifically Trifluoroacetic Acid, TFA), which is utilized at the final stage of synthesis.
2. The SPPS Cycle (Step-by-Step)
The synthesis occurs through a continuous repetition of three primary steps within the reaction vessel holding the solvated resin.
Step A: Deprotection: The cycle begins by exposing the resin-bound peptide (or the initial resin-linked amino acid) to a basic solution (20% piperidine in DMF). This base triggers an elimination reaction, rapidly cleaving the Fmoc protecting group from the N-terminus, exposing a highly reactive primary amine, and liberating a stable dibenzofulvene byproduct. The resin is then thoroughly washed with DMF to remove all traces of piperidine.
Step B: Activation and Coupling: The incoming, Fmoc-protected amino acid must be chemically activated at its carboxyl group to form an ester intermediate specifically capable of reacting with the newly exposed amine on the resin. This cannot occur spontaneously at room temperature. The amino acid is pre-mixed with an activating coupling reagent. Common highly efficient reagents include uronium salts (HBTU or HATU) or carbodiimides (DIC) in conjunction with additives like OxymaPure or HOBt to suppress unwanted racemization (loss of stereochemical chirality). A tertiary amine base, such as Diisopropylethylamine (DIEA), is added to initiate the reaction. This potent, activated amino acid slurry is pushed into the resin bed. The activated carboxyl group performs a nucleophilic attack on the exposed amine of the resin-bound peptide, forming a new, stable covalent amide (peptide) bond.
Step C: Washing and Capping (Optional but Recommended): The resin is rigorously washed with DMF to flush out unreacted amino acids and spent coupling reagents. To prevent any unreacted amines (where coupling failed, perhaps due to steric hindrance) from participating in subsequent cycles and creating truncated deletion peptides, a “capping” step is often employed using acetic anhydride. This acetylates any free amines, permanently terminating those specific chains.
This Deprotection-Coupling-Wash cycle is repeated iteratively, building the peptide sequence molecule by molecule, until the desired primary structure is completed.
Global Deprotection and Cleavage from the Resin
Once the final N-terminal Fmoc group is removed, the fully elongated, protected peptide remains attached to the solid resin. The critical final chemical step involves the “global” removal of all side-chain protecting groups and the simultaneous cleavage of the peptide from the resin link.
This is executed in a highly controlled environment using strong acid, predominantly concentrated Trifluoroacetic Acid (TFA, $>90%$). Because the cleavage generates highly reactive cationic carbocations from the departing tBu and Trt protecting groups (which could rapidly re-attach to the peptide and alkylate sensitive residues like tryptophan or methionine), chemical “scavengers” must be included in the TFA cocktail. Common potent scavengers include water, Triisopropylsilane (TIS), and ethanedithiol (EDT). The resin is incubated in this acidic cleavage cocktail (often called ‘Reagent K’) for 2 to 4 hours. The mixture is then rapidly filtered (leaving the empty resin behind), and the solution is aggressively precipitated using ice-cold diethyl ether. The target peptide precipitates as an amorphous white solid, while the scavenged byproducts and TFA remain highly soluble in the ether layer and are decanted away.
Downstream Processing: Purification and Analytical Validation
The resulting crude white peptide powder is rarely pure enough for rigorous in-vitro application, often containing traces of deletion sequences, stereoisomers, or incompletely deprotected fragments. Subsequent purification is mandatory.
Preparative HPLC
The primary workhorse for peptide purification is Preparative Reversed-Phase High-Performance Liquid Chromatography (Prep-RP-HPLC). The crude peptide is dissolved and loaded onto a large, high-capacity $C_{18}$ or $C_8$ column. Using complex aqueous-organic gradients (typically Water/Acetonitrile with 0.1% TFA to maintain ionization and sharp peak shape), the full-length target peptide is baseline-separated from early-eluting truncated impurities and late-eluting hydrophobic aggregates. The desired peak is detected via UV absorption (214 nm) and fractionally collected.
Lyophilization
The highly purified fractions containing pure peptide in a highly dilute water/acetonitrile/TFA solution are rapidly frozen (often using dry ice/acetone baths or liquid nitrogen) and subjected to a hard vacuum in a freeze-dryer (lyophilizer). This process of sustained sublimation removes all volatile solvents, yielding a dry, highly stable, purified peptide salt (usually a trifluoroacetate salt, though acetate or chloride salts can be prepared via ion-exchange).
Mass Spectrometry (LC-MS) Validation
Before release for research use, the final purified peptide must undergo definitive structural validation. High-Resolution Mass Spectrometry (HRMS), often coupled with analytical HPLC, is strictly employed. Electrospray Ionization (ESI) is used to verify that the monoisotopic mass of the synthesized molecule perfectly matches the theoretical calculated mass of the sequence, confirming unambiguous molecular identity and confirming the structural integrity of the synthesized product.
Conclusion
Solid-Phase Peptide Synthesis represents a triumph of automated organic chemistry. By exploiting the orthogonal reactivity of Fmoc/tBu protecting groups and the physical separation provided by insoluble resin matrices, massive and intricate molecular architectures can be assembled rapidly with staggering precision. Supported by high-resolution preparative chromatography and precise mass spectrometry, modern peptide synthesis provides the highly pure, highly validated structural ligands essential for driving specialized in-vitro biochemical analysis and modern pharmacological discovery.