
—
Cellular Immunomodulation: In-Vitro Transduction Pathways of the KPV Tripeptide in Inflammatory Models
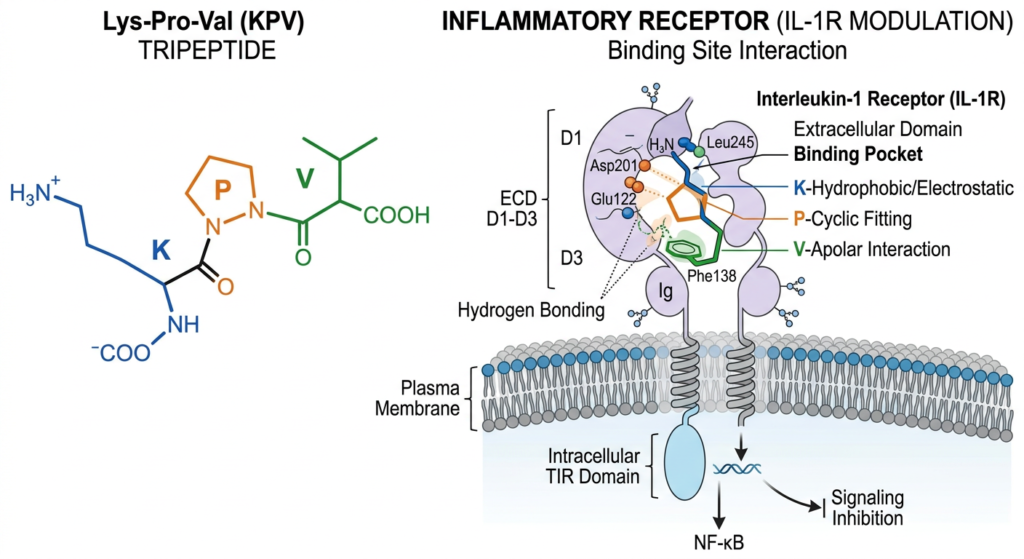
Introduction to the KPV Sequence
The tripeptide sequence Lysine-Proline-Valine (KPV) represents the highly conserved, extreme C-terminal functional sequence of the larger endogenous precursor hormone, $alpha$-melanocyte-stimulating hormone ($alpha$-MSH). While the full-length 13-amino-acid $alpha$-MSH molecule exerts profound systemic effects—modulating aggressive pigmentation, complex metabolic homeostasis, and regulating broad anti-inflammatory responses—exhaustive research has focused on isolating specific, truncated functional domains. The KPV sequence has emerged as an intensive focal point for in-vitro pharmacological investigation. It is theorized to retain a significant proportion of the potent parent molecule’s anti-inflammatory and immunomodulatory signaling capacity without heavily triggering complex systemic melanogenesis (pigmentation) or engaging central metabolic regulatory receptor pathways. This detailed review examines the specific in-vitro mechanisms, distinct receptor interactions, and the complex intracellular transduction cascades modulated by synthetic KPV in rigorous cellular models of inflammation.
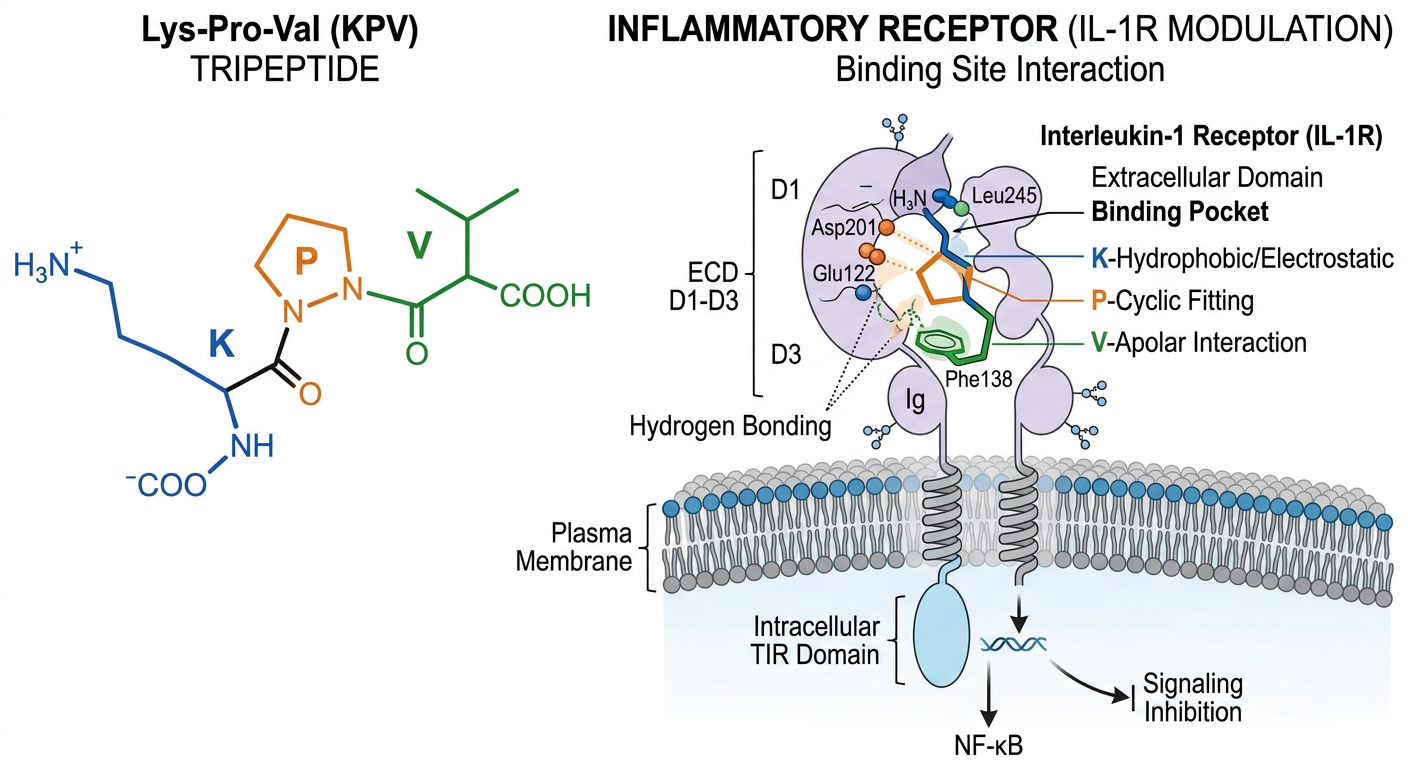
Beyond the GPCR: Investigating Receptor Targets
The classic mechanism of action for full-length melanocortin peptides like $alpha$-MSH is binding to and activating the five established G-protein-coupled melanocortin receptors (MC1R-MC5R). These classic receptors reside on the cell membrane surface and rapidly activate the massive intracellular cAMP signaling cascade upon agonist binding.
However, the specific pharmacology of the significantly truncated KPV tripeptide remains a subject of intense and complex in-vitro debate and ongoing analysis. Because it comprises only three amino acids, it lacks the vital His-Phe-Arg-Trp conserved core sequence necessary for strong, high-affinity binding to the deep transmembrane pockets of the classic melanocortin receptors.
While some specialized in-vitro radioligand binding assays analyzing specific over-expressed cell lines suggest that KPV can still interact, albeit with significantly lower binding affinity, with the peripheral inflammatory receptor MC1R, subsequent cellular evidence strongly indicates that KPV’s primary powerful anti-inflammatory actions are likely achieved through alternative, non-classical melanocortin receptor-independent mechanisms.
Recent intensive cellular research focuses firmly on KPV’s interaction within the complex intestinal tract system. Dedicated in-vitro permeability assays utilizing sophisticated, polarized human intestinal epithelial cellular monolayers (such as Caco-2 models) have demonstrated a remarkable phenomenon: the highly water-soluble KPV peptide is capable of rapid, massive cellular internalization. Specialized membrane transporters—specifically the vast mammalian proton-coupled oligopeptide transporter (POT) family, primarily PepT1, which is heavily expressed on the apical surface of intestinal enterocytes—rapidly internalize the tripeptide structure deep into the cellular cytosol. This robust intracellular penetration represents a radical paradigm shift; it suggests KPV does not simply bind completely on the external cellular membrane but actively executes its anti-inflammatory signaling from within the highly complex intracellular environment.
The NF-$kappa$B Inhibition Cascade
The primary and most widely documented biochemical mechanism underlying KPV’s robust anti-inflammatory capability, heavily observed across numerous diverse in-vitro cellular arrays (from human intestinal epithelial cells to aggressively stimulated macrophage colonies), is its profound capacity to heavily disrupt the master inflammatory signaling pathway: the Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-$kappa$B) cascade.
Under normal, highly inflammatory conditions—such as cellular exposure to massive pathogenic stimuli like lipopolysaccharide (LPS, representing bacterial endotoxin) or the rapid introduction of highly potent pro-inflammatory cytokines like TNF-$alpha$ or aggressive Interleukin-1$beta$ (IL-1$beta$)—a massive intracellular complex of specific kinases (the IKK complex) is rapidly phosphorylated and aggressively activated. This IKK complex then immediately phosphorylates I$kappa$B, the vital inhibitory protein complex that tightly binds and paralyzes the NF-$kappa$B dimer, holding it inactive within the cellular cytosol. Once I$kappa$B is structurally phosphorylated, it is rapidly flagged for devastating breakdown and complete cellular degradation by the massive proteasome complex.
With its inhibitor completely destroyed, the potent active NF-$kappa$B dimer is violently liberated. It immediately and rapidly forcefully translocates straight into the cellular nucleus. There, it tightly binds to highly specific target DNA promoter sequences, initiating completely massive, rapid, and aggressive genetic transcription and immediate subsequent massive cellular production of numerous massive inflammatory mediators, specifically heavy up-regulation of COX-2 enzymes and massive cellular secretion of inflammatory cytokines (IL-6, IL-8, TNF-$alpha$).
Rigorous analytical and complex in-vitro biochemical assays (such as cellular Western blotting to track complex protein locations and rapid electrophoretic mobility shift assays, EMSA, to directly visualize active DNA binding interactions) powerfully demonstrate that the direct, heavy application of highly concentrated KPV directly, firmly intercept this master pathway. Specifically, powerful intra-cellular KPV acts to totally prevent the critical phosphorylation and subsequently catastrophic degradation of the protective I$kappa$B inhibitory protein complex. By effectively stalling the destructive IKK kinases and forcing I$kappa$B to remain totally intact and functional, KPV powerfully forces the potent NF-$kappa$B dimer to stay fully immobilized and completely paralyzed within the cytosol. The critical nuclear translocation phase is strictly and heavily blocked, totally halting the massive inflammatory genetic transcription process before it can aggressively initiate.
Anti-Inflammatory Cytokine Profiling
The immediate downstream consequence of strictly disrupting the massive NF-$kappa$B signaling network is clearly visible via rigorous in-vitro cellular secretion analysis. Using rapid, highly dedicated enzyme-linked immunosorbent assays (ELISA) to rigorously quantify protein output into surrounding cellular culture media, studies heavily analyzing macrophages aggressively stimulated heavily with inflammatory bacterial LPS demonstrate a rapid, massive, and powerful dose-dependent effect upon heavy application of concentrated KPV.
Specifically, KPV acts to powerfully and massively suppress the rapid cellular secretion of highly pro-inflammatory, massively destructive cytokines, predominantly completely suppressing the release of TNF-$alpha$ and dramatically minimizing aggressive levels of IL-6. This rapid inflammatory blockade creates a heavy cellular environment entirely hostile to vast inflammation while aggressively leaving normal routine cellular homeostatic operations functionally intact and undisrupted.
Modulation of Cellular Adhesion
Beyond directly and powerfully stopping massive cytokine release, complex in-vitro analytical evaluation also heavily underscores KPV’s powerful critical role in rapidly mitigating and limiting severe inflammatory cellular recruitment cascades into vital delicate tissues.
During heavy local aggressive inflammatory responses, particularly within the sensitive endothelial lining of complex blood vessels or within severe structural intestinal lesions, local traumatized cells rapidly drastically up-regulate the heavy surface expression of large complex cellular adhesion molecules—notably Intercellular Adhesion Molecule-1 (ICAM-1) and massive Vascular Cell Adhesion Molecule-1 (VCAM-1). These massive structural surface proteins essentially function as a physical cellular anchor system, forcefully grabbing circulating protective immune cells (specifically neutrophils and heavily active macrophages) directly from the circulating blood stream and aggressively facilitating their massive, forced, rapid destructive migration directly into the soft delicate inflamed structural tissue, frequently massively worsening the initial physical tissue damage.
Specific, complex in-vitro cellular assays using directly stimulated delicate human umbilical vein endothelial cell (HUVEC) arrays powerfully show that prior or concurrent rapid heavy application of concentrated KPV powerfully down-regulates and aggressively minimizes the rapid surface expression of these critical massive adhesion molecules. By actively aggressively smoothing the inflamed cellular surface and drastically removing the physical molecular anchor points, KPV actively physically limits the massive destructive infiltration of highly reactive immune cells into damaged structural tissues.
Conclusion
The structural tripeptide KPV represents a distinct highly focused, aggressively powerful mechanism in targeted specialized cellular inflammatory modulation. Its specific analytical in-vitro pharmacological profile diverges strongly from the widespread potent receptor-mediated actions of its massive full-length precursor, $alpha$-MSH. By utilizing active rapid dedicated massive specific cellular internalization systems, KPV rapidly functions as a heavily potent intracellular biological signaling agent, directly and aggressively intercepting massive destructive cellular inflammatory cascades—specifically powerfully stalling the aggressive NF-$kappa$B cascade and immediately physically suppressing massive structural cellular adhesion networks. While rigorous precise exact in-vitro characterizations regarding specific novel internal receptor targets violently continue, KPV’s intense ability to deeply penetrate protective cells and powerfully alter massive internal destructive cellular signaling cascades defines it as a highly critical synthetic biological structure for rigorous specific continued fundamental immunopharmacological structural research.